
We are excited to share our first big milestone in solving a grand challenge that has hampered the predictive power of computational chemistry, biochemistry, and materials science for decades. By using a scalable deep-learning approach and generating an unprecedented quantity of diverse, highly accurate data, we have achieved a breakthrough in the accuracy of density functional theory (DFT), the workhorse method that thousands of scientists use every year to simulate matter at the atomistic level. Within the region of chemical space represented in our large training dataset, our model reaches the accuracy required to reliably predict experimental outcomes, as assessed on the well-known benchmark dataset W4-17 (opens in new tab). This removes a fundamental barrier to shifting the balance of molecule and material design from being driven by laboratory experiments to being driven by computational simulations. The implications for accelerating scientific discovery are far reaching, spanning applications from drugs to batteries and green fertilizers.
What is DFT?
Molecules and materials are made of atoms, which are held together by their electrons. These electrons act as a glue, determining the stability and properties of the chemical structure. Accurately computing the strength and properties of the electron glue is essential for predicting whether a chemical reaction will proceed, whether a candidate drug molecule will bind to its target protein, whether a material is suitable for carbon capture, or if a flow battery can be optimized for renewable energy storage. Unfortunately, a brute-force approach amounts to solving the many-electron Schrödinger equation, which requires computation that scales exponentially with the number of electrons. Considering that an atom has dozens of electrons, and that molecules and materials have large numbers of atoms, we could easily end up waiting the age of the universe to complete our computation unless we restrict our attention to small systems with only a few atoms.
DFT, introduced by Walter Kohn and collaborators in 1964-1965, was a true scientific breakthrough, earning Kohn the Nobel Prize in Chemistry in 1998. DFT provides an extraordinary reduction in the computational cost of calculating the electron glue in an exact manner, from exponential to cubic, making it possible to perform calculations of practical value within seconds to hours.
What is the grand challenge in DFT?
But there is a catch: the exact reformulation has a small but crucial term—the exchange-correlation (XC) functional—which Kohn proved is universal (i.e., the same for all molecules and materials), but for which no explicit expression is known. For 60 years, people have designed practical approximations for the XC functional. The magazine Science dubbed the gold rush to design better XC models the “pursuit of the Divine Functional (opens in new tab)”. With time, these approximations have grown into a zoo of hundreds of different XC functionals from which users must choose, often using experimental data as a guide. Owing to the uniquely favorable computational cost of DFT, existing functionals have enabled scientists to gain extremely useful insight into a huge variety of chemical problems. However, the limited accuracy and scope of current XC functionals mean that DFT is still mostly used to interpret experimental results rather than predict them.
Why is it important to increase the accuracy of DFT?
We can contrast the present state of computational chemistry with the state of aircraft engineering and design. Thanks to predictive simulations, aeronautical engineers no longer need to build and test thousands of prototypes to identify one viable design. However, this is exactly what we currently must do in molecular and materials sciences. We send thousands of potential candidates to the lab, because the accuracy of the computational methods is not sufficient to predict the experiments. To make a significant shift in the balance from laboratory to in silico experiments, we need to remove the fundamental bottleneck of the insufficient accuracy of present XC functionals. This amounts to bringing the error of DFT calculations with respect to experiments within chemical accuracy, which is around 1 kcal/mol for most chemical processes. Present approximations typically have errors that are 3 to 30 times larger.
How can AI make a difference?
AI can transform how we model molecules and materials with DFT by learning the XC functional directly from highly accurate data. The goal is to learn how the XC functional captures the complex relationship between its input, the electron density, and its output, the XC energy. You can think of the density like a glue, with regions of space where there is a lot of it and other regions with less of it. Traditionally, researchers have built XC functional approximations using the concept of the so-called Jacob’s ladder: a hierarchy of increasingly complex, hand-designed descriptors of the electron density. Including density descriptors from higher rungs of this ladder aims to improve accuracy, but it comes at the price of increased computational cost. Even the few attempts that use machine learning have stayed within this traditional paradigm, thereby taking an approach that is akin to what people were doing in computer vision and speech recognition before the deep-learning era. Progress toward better accuracy has stagnated for at least two decades with this approach.
Our project is driven by the intuition that a true deep learning approach—where relevant representations of the electron density are learned directly from data in a computationally scalable way—has the potential to revolutionize the accuracy of DFT, much like deep learning has transformed other fields. A significant challenge with going down this path, however, is that feature or representation learning is very data-hungry, and there is very little data around—too little to test this hypothesis reliably.
What have we done in this milestone?
The first step was generating data—a lot of it. This posed a major challenge, since the data must come from accurate solutions of the many-electron Schrödinger equation, which is precisely the prohibitively expensive problem that DFT is designed to replace. Fortunately, decades of progress in the scientific community have led to smarter, more efficient variants of brute-force methods, making it possible to compute reference data for small molecules at experimental accuracy. While these high-accuracy methods, also referred to as wavefunction methods, are far too costly for routine use in applications, we made a deliberate investment in them for this project. The reason? The upfront cost of generating high-quality training data is offset by the long-term benefit of enabling vast numbers of industrially relevant applications with cost effective DFT using the trained XC functional. Crucially, we rely on the ability of DFT—and our learned XC functional—to generalize from high-accuracy data for small systems to larger, more complex molecules.
There are many different high-accuracy wavefunction methods, each tailored to different regions of chemical space. However, their use at scale is not well established, as they require extensive expertise—small methodological choices can significantly affect accuracy at the level that we target. We therefore joined forces with Prof. Amir Karton (opens in new tab) from the University of New England, Australia, a world-leading expert who developed widely recognized benchmark datasets for a fundamental thermochemical property: atomization energy—the energy required to break all bonds in a molecule and separate it into individual atoms. To create a training dataset of atomization energies at unprecedented scale, our team at Microsoft built a scalable pipeline to produce highly diverse molecular structures. Using these structures and substantial Azure compute resources via Microsoft’s Accelerating Foundation Models Research program (opens in new tab), Prof. Karton applied a high-accuracy wavefunction method to compute the corresponding energy labels. The result is a dataset (opens in new tab) two orders of magnitude larger than previous efforts. We are releasing a large part of this dataset (opens in new tab) to the scientific community.
Data generation was only half of the challenge. We also needed to design a dedicated deep-learning architecture for the XC functional—one that is both computationally scalable and capable of learning meaningful representations from electron densities to accurately predict the XC energy. Our team of machine learning specialists, assisted by DFT experts, introduced a series of innovations that solve these and other challenges inherent to this complex learning problem. The result is Skala, an XC functional that generalizes to unseen molecules, reaching the accuracy needed to predict experiments. This demonstrates for the first time that deep learning can truly disrupt DFT: reaching experimental accuracy does not require the computationally expensive hand-designed features of Jacob’s ladder. Instead, we can retain the original computational complexity of DFT while allowing the XC functional to learn how to extract meaningful features and predict accurate energies.
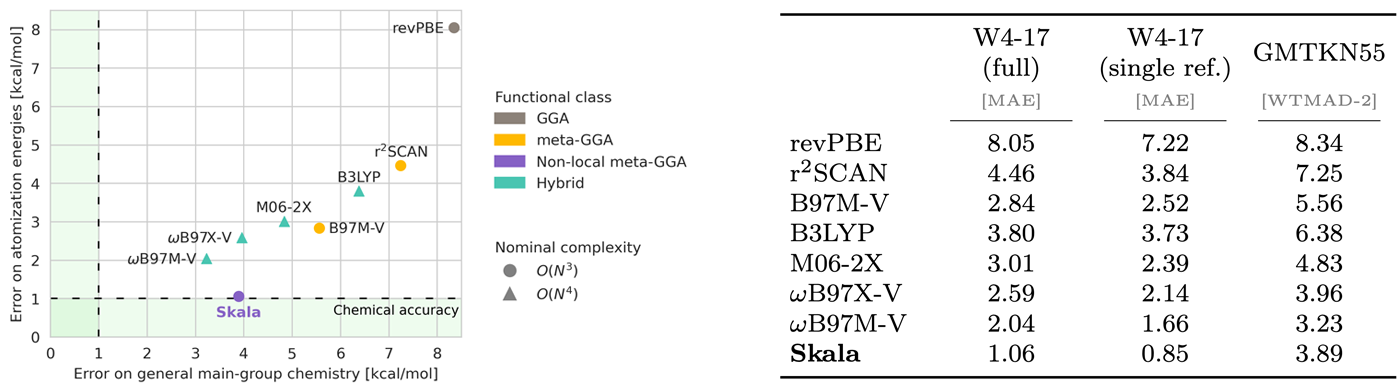
“Skala is a new density functional for the exchange-correlation energy that employs meta-GGA ingredients plus D3 dispersion and machine-learned nonlocal features of the electron density. Some exact constraints were imposed, and some others “emerge” from the fitting to about 150,000 accurate energy differences for sp molecules and atoms. Skala achieves high, hybrid-like accuracy on a large and diverse data set of properties of main group molecules, which has no overlap with its training set. The computational cost of Skala is higher than that of the r2SCAN meta-GGA for small molecules, but about the same for systems with 1,000 or more occupied orbitals. Its cost seems to be only 10% of the cost of standard hybrids and 1% of the cost of local hybrids. Developed by a Microsoft team of density functional theorists and deep-learning experts, Skala could be the first machine-learned density functional to compete with existing functionals for wide use in computational chemistry, and a sign of things to come in that and related fields. Skala learned from big data and was taught by insightful human scientists.”
— John P. Perdew, Professor of Physics, School of Science and Engineering, Tulane University
This first milestone was achieved for a challenging property in a specific region of chemical space—atomization energies of main group molecules—for which we generated our initial large batch of high-accuracy training data. Building on this foundation, we have started to expand our training dataset to cover a broader range of general chemistry, using our scalable in-house data generation pipeline. With the first small batch of training data beyond atomization energies, we have already extended the accuracy of our model, making it competitive with the best existing XC functionals across a wider spectrum of main group chemistry. This motivates us to continue growing our high-accuracy data generation campaign, engaging with external experts such as Prof. Amir Karton, who noted, “After years of benchmarking DFT methods against experimental accuracy, this is the first time I’ve witnessed such an unprecedented leap in the accuracy–cost trade-off. It is genuinely exciting to see how the creation of our new dataset has enabled these groundbreaking results — opening up a path for transformative advances across chemical, biochemical, and materials research.”
Advancing computational chemistry together
We are excited to work closely with the global computational chemistry community to accelerate progress for all and look forward to openly releasing our first XC functional in the near future.
“Density Functional Theory (DFT) and related technologies are a core Digital Chemistry technology supporting advancements in Merck’s diverse Life Science, Healthcare and Electronics businesses. However, the limitations of traditional DFT methods, which have persisted for the last 50 years, have hindered its full potential. Microsoft Research’s innovative approach to integrating deep learning represents a substantial leap, enhancing its accuracy, robustness, and scalability. We are looking forward to exploring how this can advance Digital Chemistry workflows and unlock new possibilities for the future, aligning with our commitment to developing advanced algorithms and technologies that propel scientific innovation at Merck.”
— Jan Gerit Brandenburg – Director for Digital Chemistry at Merck
“We are entering a golden age for predictive and realistic simulations: very accurate electronic-structure calculations provide vast amounts of consistent data that can be used to train novel machine-learning architectures, delivering the holy grail of precision and computational efficiency.”
— Professor Nicola Marzari, Chair of Theory and Simulation of Materials, EPFL and PSI
We believe that our new functional can help unlock new opportunities for businesses and are eager to work together on real-world applications. Today, we are delighted to launch the DFT Research Early Access Program (DFT REAP) and welcome Flagship Pioneering as the first participant. This program is for companies and research labs to collaborate with us to accelerate innovation across many industries. To find out more about how to join this program please visit: https://aka.ms/DFT-REAP (opens in new tab)
“Microsoft’s effort to enhance the predictive power of computational chemistry reflects a bold but thoughtful step toward a simulation-first future. At Flagship, we believe that openly shared, foundational advances in science – like this leap forward in DFT accuracy – can serve as powerful enablers of innovation. These next-generation tools promise to accelerate discovery across a wide range of sectors, from therapeutics to materials science, by helping researchers navigate chemical and biological space with far greater precision and speed.”
— Junaid Bajwa, M.D., Senior Partner at Flagship Pioneering and Science Partner at Pioneering Intelligence
By making our work available to the scientific community, we hope to enable widespread testing and gather valuable feedback that will guide future improvements. For the first time, deep learning offers a clear and computationally scalable path to building an accurate, efficient, and broadly applicable model of the universal XC functional—one that could transform the computational design of molecules and materials.
Acknowledgement
This work is the product of a highly collaborative and interdisciplinary effort led by Microsoft Research AI for Science, in partnership with colleagues from Microsoft Research Accelerator, Microsoft Quantum and the University of New England. The full author list includes Giulia Luise, Chin-Wei Huang, Thijs Vogels, Derk P. Kooi, Sebastian Ehlert, Stephanie Lanius, Klaas J. H. Giesbertz, Amir Karton, Deniz Gunceler, Megan Stanley, Wessel P. Bruinsma, Victor Garcia Satorras, Marwin Segler, Kenji Takeda, Lin Huang, Xinran Wei, José Garrido Torres, Albert Katbashev, Rodrigo Chavez Zavaleta, Bálint Máté, Sékou-Oumar Kaba, Roberto Sordillo, Yingrong Chen, David B. Williams-Young, Christopher M. Bishop, Jan Hermann, Rianne van den Berg and Paola Gori Giorgi.

















